Questo gruppo di uveiti si manifesta con lesioni della coroide e del complesso EPR-coriocapillare. Alcune sono patologie infiammatorie, altre patologie infettive e quasi sempre associate ad altre manifestazioni oculari ed extraoculari. I processi infiammatori che interessano la retina difficilmente rimangono localizzati nel tessuto primariamente affetto ma diffondono nelle strutture vicine determinando un’uveo-retinite. Si parla pertanto di retino-coroidite quando una primitiva retinite interessa secondariamente la sottostante coroide e viceversa di corio-retinite quando una primitiva flogosi della coroide interessa secondariamente la retina.
Secondo la più attuale classificazione proposta dal SUN le uveiti posteriori possono essere divise in:
-
Coroiditi focali, multifocali, diffuse
-
Corioretiniti
-
Retinocoroiditi
-
Retiniti
-
Neuroretiniti
Un modo schematico per approcciarsi alla diagnosi di uveite posteriore può essere la suddivisione delle manifestazioni cliniche oculari in base alla localizzazione della lesione o dell’infiammazione:
1. U. posteriori con coinvolgimento predominante del vitreo
2. U. posteriori con coinvolgimento predominante della retina superficiale
3. U. posteriori con coinvolgimento predominante della retina profonda e/o della coroide con aspetto focale o multifocale
4.
Neuroretiniti
1. Uveiti posteriori con coinvolgimento predominante del vitreo
Toxoplasmosi
La malattia può essere congenita (trasmissione transplacentare) con localizzazione oculare solitamente bilaterale o acquisita per ingestione di oocisti o cisti tessutali (trasmissione oro-fecale).
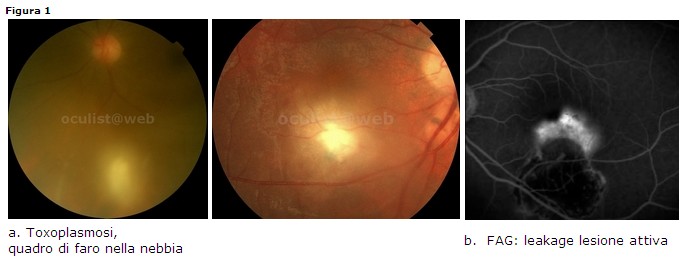
Clinica: la reazione cellulare vitreale sovrastante il focolaio retinico (solitamente satellite di una lesione cicatriziale) può essere molto marcata (quadro di faro nella nebbia) (Fig. 1a). L’assenza di cellule infiammatorie nel vitreo posteriore è sempre un elemento di esclusione nella diagnosi di retinocoroidite toxoplasmica in fase attiva.
Diagnosi:
· Sierologia con valore limitato, possibile la ricerca anticorpi in umor acqueo (elevata sensibilità) e/o l’impiego della PCR (alta specificità).
· FAG: leakage a livello del focolaio attivo, ipofluorescenze satelliti (75%) della lesione attiva (Fig. 1b).
Candidosi
Tossicodipendenza, AIDS, terapia sistemica prolungata, nutrizione parenterale
sono le condizioni più comuni che predispongono ad infezioni micotiche
opportunistiche. Infezioni esogene possono svilupparsi dopo chirurgia
intraoculare ma più rare e meno aggressive rispetto alle forme endogene [32].
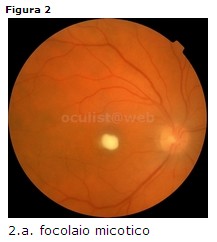
Clinica: monolaterale/bilaterale, lesioni bianco-giallastre cotonose, che
attraversano la limitante interna e invadono la camera vitrea. Inizialmente i
mezzi diottrici sono limpidi ma l’evoluzione verso l’endoftalmite è la regola (Fig.
2).
Diagnosi: prevalentemente anamnestico-clinica; vitrectomia diagnostica con
striscio e/o coltura vitreale.
Toxocariasi
Colpisce soprattutto giovani con un range compreso tra i 2 e i 30 anni [33].
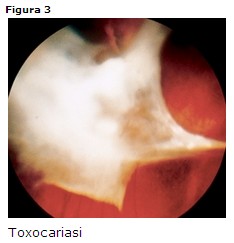
Clinica: endoftalmite, granuloma corioretinico maculare o posteriore; granuloma
corioretinico periferico con trazioni vitreo-retiniche (Fig. 3).
Diagnosi:
• Sierologia (titolo positivo >1:32), aumento IgE, ipereosinofilia, ricerca
anticorpi in UA/vitreo con test ELISA [34].
Sindrome di Whipple
E’ caratterizzata da coinvolgimento intestinale e neurologico, artrite
monoarticolare e linfadenopatia.
Clinica: opacità vitreali (tipo “more bianche”), vasculite e papilledema.
Diagnosi: VTK diagnostica, biopsia intestinale.
2. Coinvolgimento principale della retina superficiale
Toxoplasmosi
Vedi sopra.
Endoftalmite da Candida
Vedi sopra
Malattia di Behçet
Colpisce prevalentemente giovani adulti tra la 3°-4° decade; M>F; più diffusa
nel bacino del Mediterraneo e Giappone.
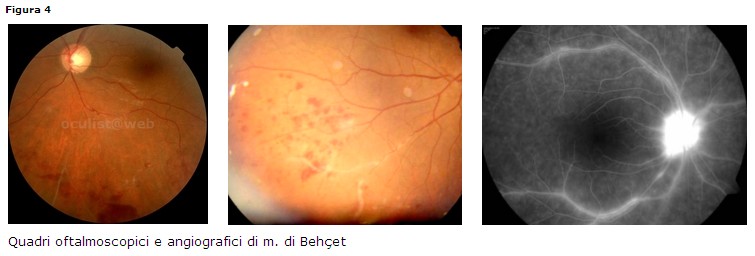
Clinica: afte orali (98%), afte genitali, disturbi articolari, disturbi
vascolari (tromboflebiti, occlusioni), disturbi neurologici. Le manifestazioni
oculari posteriori caratteristiche comprendono vasculite occlusiva (89%) venosa
e arteriosa con emorragie ed essudati, coroidite, corioretinite. Può associarsi
a diffusa vitreite (Fig. 4).
Diagnosi:
• HLA-B51
• FAG: vasculite retinica e leakage papillare e peripapillare (negli stadi
precoci della malattia)
Retinite da Herpes (ARN, Acute Retinal Necrosis)
Quadro clinico drammatico che colpisce adulti con stato immunitario indifferente
(interessa anche immunocompetenti), diversamente dalla PORN dove i pazienti sono
quasi sempre immunodepressi [35].
Clinica: focolai giallo-bianchi profondi a margini netti, tipicamente periferici
negli stadi iniziali (Fig. 16) e che confluiscono progressivamente verso il polo
posteriore; vasculite occlusiva essudativo-emorragica. Si può associare
un’importante reazione infiammatoria granulomatosa in camera anteriore con
ipertono ed una vitreite da moderata a severa. E’ frequente un distacco retinico
essudativo dopo 4-6 settimane dall’esordio della malattia.
Diagnosi: gli esami supportano la diagnosi che è prevalentemente clinica. Utile
la PCR sull’umore acqueo per la ricerca di HZV, HSV 1-2 (CMV?, EBV?) [36]. Test
HIV, sierologia sifilide.

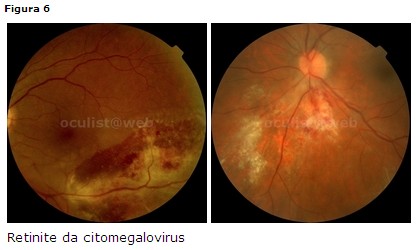
Retinite da CMV
Colpisce soggetti immunodepressi (fattore di rischio linfociti T CD4 < 50 cell/mm³,
non terapia HAART). Può essere la prima manifestazione di AIDS nel 2% dei casi
[37].
Clinica: inizia tipicamente con uno o due foci lungo i vasi con essudati
intraretinici ed emorragie sul bordo della lesione. Si può associare a vasculite
occlusiva mentre la reazione vitreale è modesta (Fig. 6). L’evoluzione delle
lesioni consiste in aree grigie inattive o cicatrici gliotiche trasparenti. In
pazienti che iniziano la HAART con preesistente retinite attiva, è possibile un
fenomeno definito IRU (“immune ricovery uveitis”) ovvero una reazione
infiammatoria importante che può essere un fenomeno transitorio o che può
protrarsi con complicanze croniche.
Diagnosi: test HIV, sierologia CMV. In casi non responsivi alla terapia si
esegue PCR su campioni biologici.
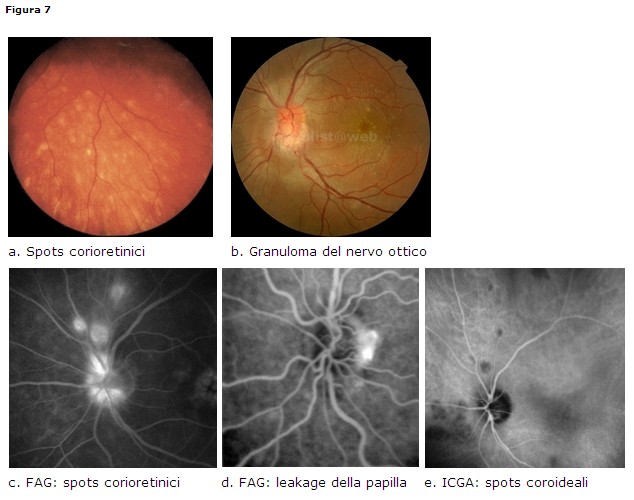
Sarcoidosi retinica
E’ possibile un esordio acuto (S. di Lofgren), subacuto, insidioso (S. di Heerfordt-Waldenstrom). Età 20-40 anni.
Clinica: le lesioni del segmento posteriore, di solito monolaterali in fase acuta, colpiscono il 25% dei pazienti [38] con sarcoidosi oculare. Le manifestazioni a livello del segmento posteriore sono: vitreite, corio-retinite con spots bianchi rotondeggianti multipli inferiori, periflebite principalmente venosa a “gocce di cera”, occlusioni vascolari, neovascolarizzazione retinica, essudati vitreali basali a palla di neve o a collana di perle (pars planite), papilledema e granuloma della testa del nervo ottico (Fig. 7).
Diagnosi:
-
ACE, lisozima, Mantoux, Rx torace, scintigrafia Ga67, biopsia del granuloma.
-
FAG + ICGA: il verde indocianina è importante per identificare lesioni coroideali subcliniche che appaiono come spots ipofluorescenti nelle fasi intermedie e diventare iso o restare ipofluorescenti nelle fasi tardive.
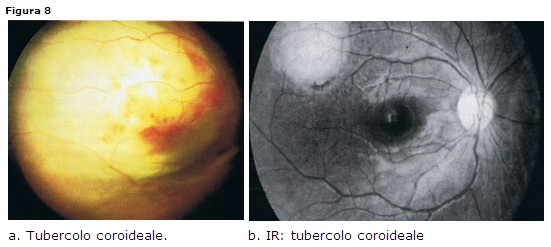
TBC retinica
Colpisce prevalentemente immunodepressi e immigrati da aree endemiche. Interessa
solo l’1.4% dei pazienti con malattia in fase attiva.
Clinica: da vitreite di basso grado fino a vitreite importante con snowballs nel
settore inferiore (pars planite); tubercoli coroideali singoli o multipli
grigio-biancastri (Fig. 8 a-b) e periflebite sia periferica sia dei grossi
vasi.
Diagnosi:
• PPD
• FAG + ICGA: l’utilizzo del verde indocianina è importante per identificare
lesioni subcliniche; le lesioni possono presentarsi come spots ipofluorescenti
nelle fasi intermedie e diventare iso o restare ipofluorescenti nelle fasi
tardive. Altri segni all’ICGA, indicanti una fase acuta, sono piccole lesioni
iperfluorescenti dette pin-points e l’iperfluorescenza coroideale tardiva [39].
3. Coinvolgimento predominante della retina profonda e/o della coroide
Retinocoroidite da Toxoplasma
Vedi sopra
Sarcoidosi
Vedi sopra
TBC
Vedi sopra
Sifilide
Sono colpiti soggetti che hanno rapporti sessuali a rischio. Si presenta con
lesioni cutanee (rash maculo-papuloso mani e piedi) e manifestazioni
neurologiche (sifilide terziaria).
Clinica: coroidite diffusa, corioretinite, vasculite (soprattutto periflebite)
ed emorragie preretiniche. Può coesistere un’importante vitreite. Evolve in
estese cicatrici corioretiniche con intensa migrazione pigmentaria.
Diagnosi:
• Clinica
• VDRL e FTA-ABS
• Richerca HIV
• Esame liquor in HIV+
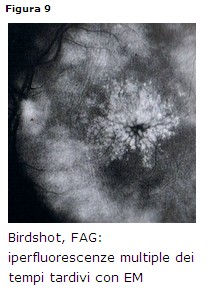
Corioretinopatia tipo “birdshot”
Età media 50 anni, vitiligine.
Clinica: solitamente bilaterale, cellularità lieve o moderata in camera vitrea,
spots corioretinici ovali color crema o depigmentati senza aree di
iperpimentazione retroequatoriali, edema maculare cistoide, restringimento
arteriolare, edema della testa del nervo ottico.
Diagnosi
• HLA-A29 (sensibilità del 96% e specificità del 93%) [40].
• FAG: si evidenzia un’ipofluorescenza precoce e un’iperfluorescenza tardiva
delle chiazzette (Fig. 9). E’ comune l’iperfluorescenza del nervo ottico.
• ICGA: lesioni ipofluorescenti precoci (per occlusione dei vasi arteriosi
coroideali), iperfluorescenti nei tempi tardivi.
• ERG: anomali
Sindrome di Vogt-Koyanagi-Harada
Interessa soggetti intorno alla 3°-4° decade, più frequentemente giapponesi e
ispano-americani e più spesso di sesso femminile. Le manifestazioni sistemiche
sono: disturbi neurologici (atassia e confusione) e uditivi, poliosi delle
ciglia, vitiligine, madarosi e alopecia negli stadi più avanzati.
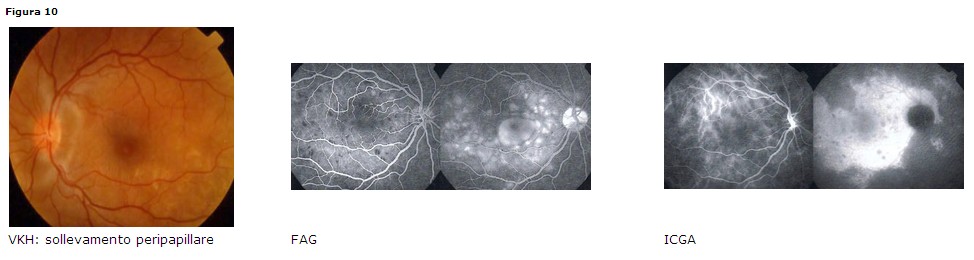
Clinica: coinvolgimento bilaterale, sollevamento corioretinico peripapillare (Fig.
10a), distacchi multipli sierosi retinici, edema della testa del nervo ottico, depigmentazione della coroide negli stadi più avanzati della malattia (fundus
con aspetto “a tramonto infuocato”) con cicatrici corioretiniche multiple.
Diagnosi:
• FAG: precoci iperfluorescenze multiple a livello dell’EPR, staining del fluido
sottoretinico nelle fasi tardive
• ICGA: ipofluorescenza intermedia e iperfluorescenza tardiva
• Esame liquor cefalorachidiano (pleiocitosi)
• Esame audiometrico
AMPPE (Acute multifocal placoid pigment epitheliopathy)
I pazienti colpiti sono generalmente giovani o di mezza età (20-50 anni), senza
predilezione di sesso. Le manifestazioni oculari sono precedute da prodromi
virali.
Clinica: lesioni spesso bilaterali placoidi giallo-crema disseminate al polo
posteriore; possono concomitare distacchi sierosi retinici. Il vitreo può essere
modestamente corpuscolato.
Diagnosi
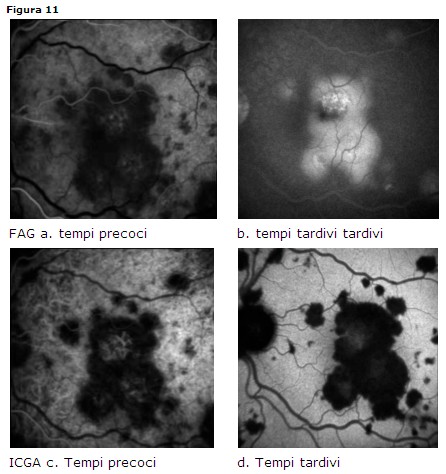
• FAG: lesioni ipofluorescenti negli stadi iniziali, iperfluorescenti negli
stadi tardivi (Fig. 11 a-b)
• ICGA: placche ipofluorescenti ai tempi precoci, intermedi e tardivi [41] e
[42] sia nella fase attiva sia nella fase cicatriziale (Fig. 11 c-d))
• ERG e EOG: variabili [43].
MEWDS (Multiple White Dot Syndrome)
La maggior parte dei pazienti è di sesso femminile, con un’età media di 30 anni.
Talvolta la malattia si associa a prodromi virali.
Clinica: monolaterale 90%, cellularità vitreale modesta, lesioni molto piccole
bianche granulari al polo posteriore interessanti la retina profonda e l’EPR.
Diagnosi:
• ERG e EOG: anomali
• FAG: spots iperfluorescenti precoci e tardivi, leakage disco ottico
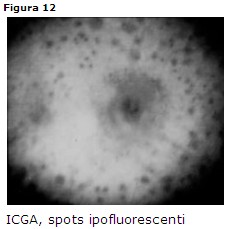
• ICGA: caratteristica ipofluorescenza precoce e tardiva (Fig. 12)
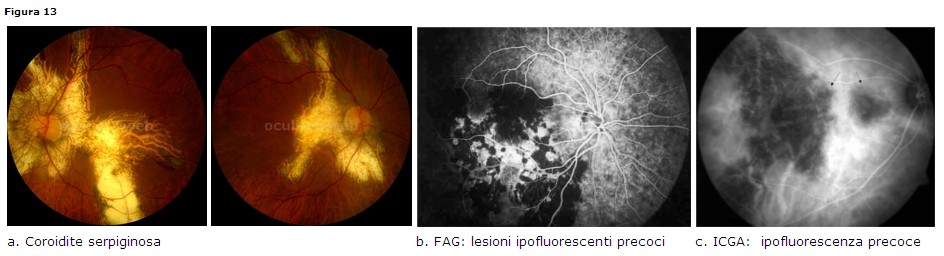
Coroidite serpiginosa o a carta geografica
I pazienti colpiti sono adulti di età compresa tra i 40 e 50 anni. Non c’è
predilezione di sesso.
Clinica: presentazione bilaterale, coinvolge coriocapillare e EPR, con lieve
cellularità vitreale; le lesioni grigiastre ed edematose partono dal nervo
ottico con andamento centrifugo, pseudopodale, in tutte le direzioni ed esitano
in cicatrici atrofiche corioretiniche.
Diagnosi
• FAG: ipofluorescenza precoce, con iperfluorescenza tardiva ai bordi della
lesione (staining) (Fig. 13 b)
• ICGA: ipofluorescenza lungo tutte le fasi dell’esame (Fig. 13 c)
PIC (Punctate inner choroidopathy)
Interessa giovani donne sane, solitamente miopi.
Clinica: bilaterale, assente cellularità vitreale, piccole opacità giallastre al
polo posteriore localizzate al di sotto dell’EPR, con evoluzione cicatriziale.
Diagnosi:
• FAG: ipofluorescenza precoce, con aumento della fluorescenza nella fasi
tardive
• ICGA: spots ipofluorescenti.
Coroidite multifocale
Colpisce donne giovani (età media 30 anni) e solitamente miopi.
Clinica: bilaterale 80%, cellularità vitreale costante, numerosi spots gialli o
grigi in regione peripapillare o in media periferia. Tali lesioni esitano in
cicatrici corioretiniche.
Diagnosi
• ERG e EOG: nella norma
• FAG: iperfluorescenza precoce dei bordi della lesione, staining tardivo della
parte centrale
• ICGA: spots ipofluorescenti (Fig. 14)
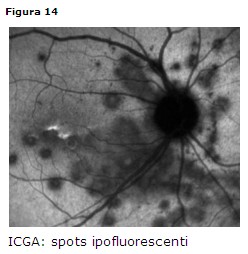
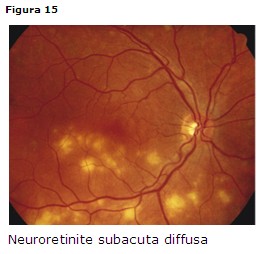
Neuroretinite diffusa subacuta monolaterale
Consegue ad infestazione da parte di nematodi ed è diffusa nel Sud-Est degli
Stati Uniti, nei Caraibi e in America latina.
Clinica: essudazione vitreale, piccoli focolai retinici profondi e periferici (Fig.
15), edema papillare.
Diagnosi:
• FAG: spots ipofluorescenti, leakage nervo ottico, staining tardivo
• ERG: anomalo
Epitelite retinica acuta
Colpisce solitamente giovani in età compresa tra 20-50 anni, in buona salute.
Clinica: piccole chiazze grigio scuro maculari; si può associare una modesta
vitreite.
Diagnosi:
• FAG: spots ipofluorescenti circondati da alone iperfluorescente
• ERG normale, EOG anomalo nella fase acuta
Riportiamo di seguito una tabella riassuntiva dei quadri principali di
uveite posteriore:
| Diagnosi differenziale nelle uveiti posteriori principali | |||||
|
PATOLOGIA |
Clinica |
CellulariTà vitreale |
Lesioni fundus |
FAG/ICGA |
Altri test |
|
APMPPE |
M=F, prodromi virali, bilaterale, giovani |
Modesta cellularità vitreale |
Lesioni larghe color crema al polo posteriore |
ICGA: Precoce ipofluo con staining tardivo |
Pleiocitosi, ERG e EOG nella norma |
|
ARN |
Stato immunitario indifferente |
Da moderata a importante |
Lesioni retiniche periferiche confluenti |
--- |
PCR |
|
Birdshot |
50 aa, vitiligine, bilaterale |
Vitreite lieve-moderata |
Foci indistinti (500-1500 micron) al polo posteriore |
Precoce ipofluo., iperfluo. tardiva, leakage maculare e del disco |
HLA-A29 (90%), ERG anomali |
|
Candida |
Immunodepressi, ospedalizzati |
Da limpido a torbido |
Lesione cotonosa al polo posteriore |
--- |
biopsia |
|
Citomegalovirus |
Immunodepressione |
Modesta vitreite |
Foci retinite con emorragie |
--- |
Test HIV, PCR |
|
Coroidite multifocale |
F>M, miopia |
Vitreite costante |
Spots giallo-grigi peripapillari |
ICGA: lesioni ipofluo. |
ERG, EOG nella norma |
|
Coroidopatia serpiginosa |
Insidioso, 50 anni, M=F, bilaterale |
Media cellularità in CA e vitreo |
Lesioni elicoidali dalla papilla |
ICGA: ipofluo. precoce, iperfluo tardiva |
--- |
|
Epitelite retinica acuta |
Acuta, giovani, M=F, unilaterale (75%) |
Quiete |
Piccoli spots neri con alone perifoveale |
Aree ipofluo circondate da alone iperfluo |
--- |
|
M. di Behçet |
3°-4° decade, M>F |
Vitreite importante |
Vasculite occlusiva, corioretinite |
FAG: vasculite, leakage papillare |
HLA-B51 |
|
MEWDS |
Esordio acuto, giovani, F>>M, unilaterale |
Media cellularità vitreale |
Lesioni granulari bianche al polo posteriore |
ICGA: ipofluo. costante |
ERG e EOG anomali |
|
PIC |
Esordio insidioso, giovani, miopi, F>>M |
Quiete |
Lesioni puntate bianche al polo posteriore (50-300 micron) |
Blocco precoce, staining tardivo |
ERG e EOG normali |
|
Sarcoidosi |
F>M |
Vitreite |
Corioretinite a chiazze multiple, periflebite, pars planite |
ICGA: Ipofluo. precoce, iperfluo. Tardiva |
ACE, lisozima |
|
Toxocariasi |
Giovani |
Importante |
Granuloma |
--- |
Eosinofilia, IgE |
|
Toxoplasmosi |
F=M |
Vitreite costante da media a severa |
Focolaio grigio-giallastro satellite di cicatrice corioretinica |
FAG: leakage lesione attiva |
Toxotest |
|
Tubercolosi |
Immunodepressione |
Vitreite da lieve a importante |
Coroidite multifocale |
ICGA: ipofluo. precoce, iperfluo. Tardiva |
PPD |
|
VHK |
F>M, 30-40 aa, bilaterale, vitiligine, disturbi neurologici. Disacusia |
Vitreite modesta |
Distacchi sierosi multipli retinici |
ICGA: precoce ipofluo., iperfluo tardiva |
HLA-A29 |
4. Neuroretiniti
Le neuroretiniti sono un gruppo eterogeneo di malattie che colpisce solitamente
individui giovani e di mezza età (6-50 anni), senza distinzione di sesso. Le
neuroretiniti possono essere di tipo idiopatico o di natura infettiva o
infiammatoria (neuroretiniti propriamente dette). La manifestazione clinica
tipica della neuroretinite, bilaterale nel 5-33% dei casi, è rappresentata
dall’edema della papilla tipicamente precoce associato a coinvolgimento
neuroretinico (stella maculare) che fa la sua comparsa anche a distanza di 1-2
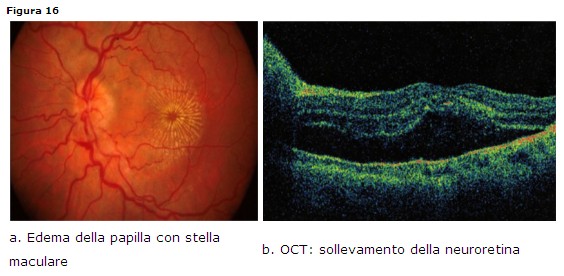
settimane e che consegue non ad un processo infiammatorio retinico ma a deposito
di materiale lipidico lungo le fibre nervose (Fig. 16a). Nel 90% dei casi è
presente cellularità vitreale. L’aspetto fluorangiografico è rappresentato da
iperfluorescenza del disco ottico, dovuta a staining, in assenza di leakage
maculare.
Le cause principali di neuroretinite sono:
• Idiopatiche
• Infettive (virali, toxoplasmosi, toxocariasi, tubercolosi, sifilide, m. di
Lyme, m. da graffio di gatto) [44] [45].
• Infiammatorie (sarcoidosi)
Dopo un’attenta valutazione degli aspetti clinico-anamnestici generali e dopo
aver escluso altre possibili cause di edema del nervo ottico con stella maculare
(malattie vascolari e tumori della testa del nervo ottico) gli esami da
richiedere sono:
• VDRL, FTA-ABS
• Sierologia Lyme
• Sierologia Bartonella
• Sierologia Toxocara
• PPD
• ACE, lisozima, Rx torace
• FAG
VASCULITI
Si intende per vasculite qualunque processo infiammatorio che coinvolga la
parete di un vaso sanguigno che spesso, ma non sempre, porta ad occlusione del
segmento interessato con possibile necrosi successiva sia del vaso sia dei
tessuti dipendenti da quel vaso. La vasculite può essere un’infiammazione
retinica vascolare primaria o, come avviene più comunemente, un interessamento
vascolare secondario ad un’infiammazione intraoculare. L’infiammazione può
coinvolgere le arterie retiniche, le vene o i capillari anche se il
coinvolgimento venoso è quello più frequentemente riscontrato. Le patologie che
si presentano con un interessamento delle arterie sono la poliarterite nodosa,
il LES e la necrosi retinica acuta, mentre le flebiti sono più spesso associate
alla malattia di Behçet, la tubercolosi, la sarcoidosi, la sclerosi multipla, la
pars planite, la malattia di Eales e le infezioni HIV correlate.
Le vasculiti retiniche possono associarsi a malattie sistemiche (sarcoidosi,
Behçet, poliartrite) oppure a malattie limitate al distretto oculare. Alcuni
casi invece esulano da questa classificazione e si parla di sindromi oculari
idiopatiche che non hanno apparenti correlazioni con altre patologie e che
vengono definite come vasculiti retiniche primitive come ad esempio la malattia
di Eales [46] e la vasculite a ramo ghiacciato.
Metodologia diagnostica nelle vasculiti
Nella valutazione di questo complesso gruppo di malattie è importante la
raccolta dei dati anamnestici ed un accurato esame clinico generale del paziente
nonostante la maggior parte dei pazienti con vasculite, al momento della
diagnosi, non presenti segni di malattia sistemica [48]. E’ necessario
riconoscere cause di tipo non infiammatorio che possono presentarsi con quadri
simili a vasculite, come la retinopatia diabetica, la retinopatia da radiazioni,
la retinopatia da emoglobinopatie, le occlusioni retiniche, la malattia di Coats
e la leucemia. A questo proposito è sicuramente utile la FAG che aiuta non solo
ad identificare la presenza di una vasculite attiva ma permette anche di
classificarla in occlusiva e non occlusiva.
Di seguito riportiamo una flow-chart utile nella diagnosi di vasculite e gli
esami che è opportuno richiedere sulla base del sospetto clinico:
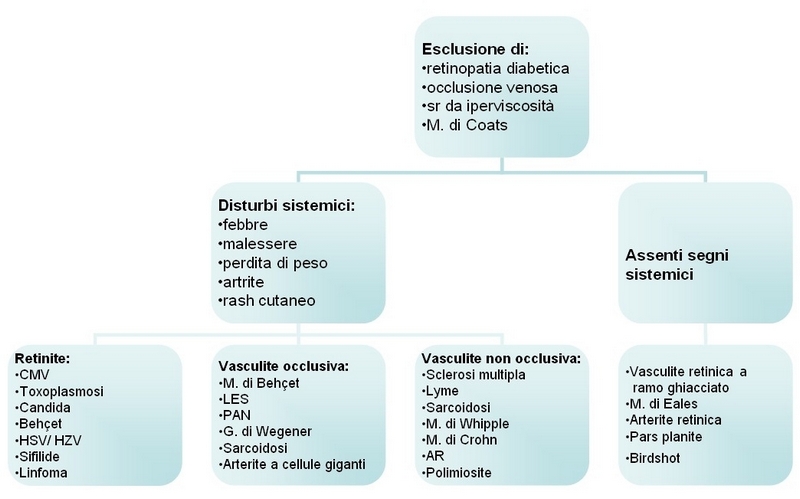
• Emocromo, esame delle urine
• CMV: test HIV, ELISA, PCR
• Toxoplasmosi: toxotest
• Candida: VTK diagnostica
• M. di Behcet: HLA-B51
• HZV/HSV: PCR dall’umor acqueo/vitreo
• Sifilide: anamnesi, VDRL, FTA-ABS
• Linfoma: RMN orbite + encefalo, VTK diagnostica, saggio IL-10, identificazione
del gene del linfoma.
• LES: ANA, anti-ds DNA, anti Sm, frazioni del complemento,
anticorpi-antifosfolipidi
• PAN: ANCA
• Granulatosi di Wegener: c-ANCA, p-ANCA
• Sarcoidosi: ACE e lisozima
• Arterite a cellule giganti: biopsia arteria temporale
• Sclerosi multipla: work-up neurologico
• M. di Lyme: sierologia.
• Malattia di Whipple: PCR per ricerca bacillo, visita internistica
• Malattia di Crohn: visita gastrointestinale
• Artrite reumatoide: FR, ANA
• Polimiosite: ANA, anti-Jo1, anti-SRP